Introduction
Many reactions are truly reversible (see blog post Part 4) and, as a
consequence, substrate(s) and product(s) are in equilibrium with each other.
The position of this equilibrium depends on thermodynamics and process conditions.
This blog post will deal with equilibriums and how to shift them more towards
the product to allow you to isolate your product in an economic manner.
There are two main ways of shifting an equilibrium towards the product
side: push and pull. Let’s look at the example in Figure 1 (a) below. This is
an ester hydrolysis in hexane as solvent where the desired molecule is the
acid. Understandably, increasing the concentration of H2O (b) will
favor hydrolysis and the arrow in the forward reaction becomes bigger. I call
that ‘pushing’ the equilibrium. We could also ‘pull’ the equilibrium towards
the product by removing the methanol by evaporation (c). We could also do both
(d). See how the arrows change?
Alternatively, we could pull the acid from the reaction medium by
binding the acid at the right pH to an anion exchange resin[1], or make it
precipitate as a salt with an appropriate counter ion, for instance.
Figure 1: Example of a lactic acid ester hydrolysis.

I will now give a few examples of solving the equilibrium issues from
enzyme classes that if you follow my blog should be familiar to you.
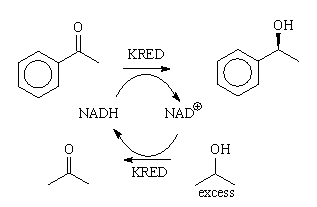
Ketoreductases
Ketone reductions are fully reversible reactions that lead to a
chemical equilibrium of ketone and alcohol (Figure 2).
Figure 2: The KRED catalyzed equilibrium reaction. [2]

If we would use a large excess of NADH, we might be able to push the
reaction to completion. However, the cost of NADH is too high to do that and,
also, high concentration of NADH acts as an inhibitor of many KREDs.
Economically, we can only use a little bit (a catalytic amount) and then only of
the cheapest form: NAD+. Therefore, the cofactor needs to be
recycled many times. This can be done by doing a similar reaction in reverse,
using a sacrificial alcohol as a co-substrate. Isopropanol (iPA) is often used
for this purpose. The scheme from Figure 2 changes to the one in Figure 3. Thus,
in the first step, the KRED oxidizes iPA to acetone and the NAD+ cofactor
is reduced to NADH. In the next step, the same KRED enzyme reduces the aryl ketone
to the chiral alcohol (hopefully) or it reduces acetone back to iPA (L).
Clearly, the efficiency of this system depends on the binding
affinities and turnover rates for iPA compared to aryl ketone. The aryl ketone
must bind better, but not so much better that it cannot be displaced by iPA
every now and then.
Figure 3: Formal scheme of KRED-catalyzed transfer hydrogenation.

Most people only use the simplified scheme shown in Figure 4 which only
becomes valid under a certain circumstance that I’ll get to in a moment. In the
mean time, note that the scheme in Figure 3 really does not have any
directional arrow. It is like the wheel of a car that can rotate in each
direction but if we want to go somewhere, it better rotate in the same
direction for a while! We need an external force that makes the wheel turn (clockwise
in this example).
That external force is created by adding a large excess of iPA: running
the enzymatic reaction in 50/50 water/iPA, or sometimes even up to 90% iPA [3]!
The large excess iPA pushes the equilibrium of the iPA oxidation all the way to
the acetone side, de facto making
that reaction uni-directional. Now, since that reaction now only goes in one
direction, the coupled reaction can also only go in one direction: towards the
chiral alcohol. We have created the scheme that is shown in Figure 4.
Figure 4: Simplied scheme for ketone reduction with excess iPA.
In some cases, the excess of iPA is not enough to push the desired
conversion above 99% yield [4, 5]. Removal of acetone by vacuum or a nitrogen
gas sweep helps pull the lower reaction to completion and thus the coupled
formation to chiral alcohol.[6] In some cases, precipitation of the chiral
alcohol can be achieved by tailoring the reaction medium; this is another case
of ‘pull’ [7].
The iPA/Acetone system is not perfect for every case: sometimes the
process conditions cannot be optimized to give adequate conversion, or the KRED
might not accept iPA as substrate. In those cases, a selection of different coupled
enzymatic reactions can be used [8]. Most popular are using glucose
dehydrogenase (GDH, [9]), formate dehydrogenase (FDH, [10]) and phosphite
dehydrogenase (PTDH, [11]) coupled systems. These reaction schemes are compiled
in Figure 5.
Figure 5: Three popular enzymatic cofactor regeneration systems.

What is important to remember in these multi-enzyme systems is that the
cofactor needs to shuttle back and forth from one enzyme to the other. This
shuttling is also something to keep in mind when KREDs are immobilized: the
fixation to the carrier must not interfere with substrate and product transport
as well as allow fast exchange of the cofactor which happens to be a rather
large molecule and (may) requires conformational changes in the protein. Apparently
it can be done since SPRIN in Italy offers a kit of 12 immobilized KREDs and
also has an immobilized GDH.[12]
Transaminases
Transaminases catalyze transfer of an amine group from a donor to a ketone and can use isopropyl amine (iPM) as amine donor, or use natural
amino acids like alanine as donor. See Figure 6 and 7 for iPM and Alanine,
respectively. Note that in these schemes, the recycle of the pyridoxal phosphate (PLP) cofactor is omitted for clarity.
Figure 6: ‘Chemical tricks’ to achieve better yield. Binding of the product-amine
to a resin also alleviates the product inhibition that is often seen with TAs.

Figure 7: Two enzymatically coupled reactions to drive transaminations
to completion. [15] And then I have not even shown another one using pyruvate
decarboxylase [16].

Haloalcohol dehalogenases
The ring closure of 1-(4-nitrophenyl)-2-bromo
ethan-1-ol (dubbed by a biochemist ‘PNSHH’ for para-nitrostyrene halo hydrin
and the name caught on...) to 4-nitro-styrene oxide (PNSO) and bromide ion was
scaled up. Reaction conditions: 15 gr substrate, 3 liters of 50% toluene/water
biphasic mixture and 100 g of acetate-loaded ion exchange resin that bound the released halide. We achieved
complete conversion with essentially absolute enantioselectivity .[17] Enzyme
load was 50 mg total and was provided by Ms Lixia Tang.
Figure 8: Since it was his idea, Jeffrey lutje Spelberg got to hold the flask in this picture which was later simply put in a shaker at 30°C.

Conclusions
Dealing with equilibrium reactions may seem daunting but as long as one
of the reactions in a coupled system is or can be made uni-directional, the
proof of concept has been made. Process optimization must follow and a rule of
thumb is “less enzyme is cheaper”. However, if using multiple enzymes can lead to a 100% yield and a simple product isolation, it can still be a winner.
References
[1] Resins available at scale, for instance, from www.purolite.com. Also look at their Purolite
LifeTech™ brand for biocatalysis and
life sciences applications.
[2] Of course this discussion does not mention NADP(H) as a cofactor
for simplicity.
[3] C Savile, JM Gruber, E Mundorff, GW Huisman, and SJ Collier. WO2010/025287
Ketoreductase polypeptides for the production of 3-aryl-3-hydroxypropanamine
from a 3-aryl-3-ketopropanamine. Patent granted to Codexis. Example 9 shows
conversion of a ketone on preparative scale at 90% iPA.
[4] J Liang, E Mundorff, R Voladri,
S Jenne, L Gilson, A Conway,
A Krebber, J Wong, GW Huisman, S Truesdell, and J Lalonde.
Highly Enantioselective Reduction of a Small Heterocyclic Ketone: Biocatalytic
Reduction of Tetrahydrothiophene-3-one to the Corresponding (R)-Alcohol. Org.
Process Res. Dev., 2010, 14 (1), pp 188–192.
[5] From an atom utilization standpoint, using ‘excess’ of something is
less elegant and frowned upon according to the 12 principles of Green Chemistry:
http://www.epa.gov/sciencematters/june2011/principles.htm
. Use of excess reagents should be avoided where possible.
[6] SJ Calvin, D Mangan, I Miskelly, TS Moody, and PJ Stevenson.
Overcoming Equilibrium Issues with Carbonyl Reductase Enzymes. Org. Process
Res. Dev. 2012, 16, 82–86. http://www.almacgroup.com/wp-content/uploads/Overcoming-Equilibrium-Issues-with-Carbonyl-Reductase-Enzymes.pdf
[7] J Liang, J Lalonde, B Borup, V Mitchell, E Mundorff, N Trinh, DA
Kochrekar, RN Cherat, and GG Pai. Development of a Biocatalytic Process as an
Alternative to the (−)-DIP-Cl-Mediated Asymmetric Reduction of a Key
Intermediate of Montelukast. Org. Process Res. Dev., 2010, 14 (1), pp 193–198.
[8] A very nice range of cofactor recycling enzymes can be acquired
from Evocatal in Germany. http://www.evocatal.com/en/products/evozymes/cofactor-regeneration.html.
Industrial scale quantities of GDH can be acquired from Codexis. FDH has decreased
in popularity because of lack of robustness under process conditions although
it was the basis for Degussa’s process for tert-Leucine. PTDH is still
experimental.
[9] See reference 4, but there are many other examples.
[10] Pioneering work was done by my friend Vladimir Tishkov from
Lomonosov University in Moscow to engineer NADP specificity in an NAD-dependent
FDH and reviewed in a nice overview: http://www.enzyme.chem.msu.ru/~tishkov/Publications/Proteins_19_FDH.pdf
[11] TW Johannes, RD
Woodyer, H Zhao. Efficient Regeneration of NADPH Using an Engineered Phosphite
Dehydrogenase. Biotech. Bioeng. 2007, 96 (1), pp 18-26. http://scs.illinois.edu/~zhaogrp/publications/HZ53.pdf
and also their nice review of cofactor recycle systems: http://www.scs.illinois.edu/~zhaogrp/publications/HZ21.pdf
[12] http://www.sprintechnologies.com
or through their US distributor: http://www.itochu-purification.com/products/biocatalysis/new-immobilized-ketoreductases
[13] K Engelmark Cassimjee, C Branneby, V Abedi, A Wells, and P Berglund.
Transaminations with isopropyl amine: equilibrium displacement with yeast
alcohol dehydrogenase coupled to in situ cofactor regeneration. Chem. Commun.,
2010, 46, pp 5569-5571.
[14] MD Truppo, JD Rozzell, and NJ Turner. Efficient Production of
Enantiomerically Pure Chiral Amines at Concentrations of 50 g/L Using
Transaminases .Organic Process Research & Development 2010, 14(1), pp.
234-237.
[15] MD Truppo, JD Rozzell, JC Moore, and NJ Turner. Rapid screening
and scale-up of transaminase catalysed reactions. Org. Biomol. Chem. 2009, 7, pp
395-398.
[16]M Höhne, S Kühl, K Robins, UT Bornscheuer. Efficient Asymmetric Synthesis of Chiral Amines by Combining
Transaminase and Pyruvate Decarboxylase. ChemBioChem 2008, 9(3), pp 363–365.
[17] JH lutje Spelberg, LX Tang, EJ de
Vries, RM Kellogg, and DB Janssen. Biocatalytic preparation of
optically pure epoxides and derivatives. Poster presentation at Biotrans 2003,
Olomouc/CZ.